Comprendre la diversité génétique de la drépanocytose
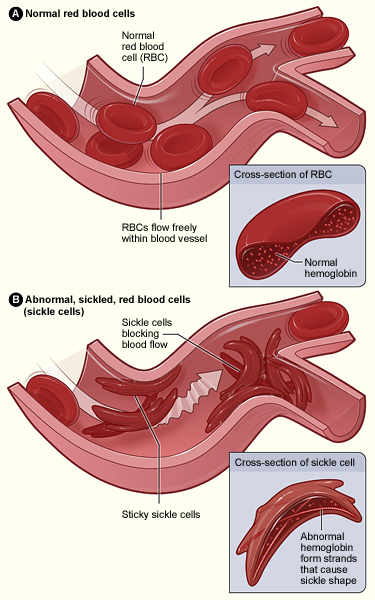
La drépanocytose n'est pas une maladie unique. Le terme recouvre en réalité plusieurs syndromes drépanocytaires majeurs, tous caractérisés par la présence d'hémoglobine S (HbS) dans les globules rouges — mais avec des combinaisons génétiques, des sévérités cliniques et des pronostics très différents.
HbSS — La forme homozygote, la plus fréquente et la plus sévère
La forme HbSS, dite drépanocytose homozygote, est la plus répandue : elle représente environ 70 % des syndromes drépanocytaires majeurs en France.
Elle survient lorsque l'enfant hérite de deux copies du gène muté codant pour l'hémoglobine S : une de chaque parent, tous deux porteurs sains (trait drépanocytaire HbAS).
Mécanisme
En situation de faible oxygénation, les molécules d'HbS polymérisent et forment des fibres rigides qui déforment le globule rouge en faucille. Ces cellules anormales :
- s'agrègent dans les petits vaisseaux → crises vaso-occlusives douloureuses
- ont une durée de vie très courte (10–20 jours vs 120 jours normalement) → anémie hémolytique chronique
- favorisent l'inflammation et les lésions vasculaires
Tableau clinique
| Complication | Fréquence |
|---|---|
| Crises douloureuses (VOC) | Très fréquentes |
| Syndrome thoracique aigu | Fréquent |
| AVC | Risque significatif (10–15 %) |
| Infections bactériennes | Risque élevé (asplénie) |
| Ulcères de jambe | Fréquents à l'âge adulte |
| Priapisme (homme) | 30–45 % des hommes |
L'espérance de vie médiane est estimée à 48 ans dans les pays à revenus élevés, contre moins de 20 ans dans les années 1970 avant les traitements modernes.
HbSC — La deuxième forme la plus fréquente
La forme HbSC représente environ 25 % des syndromes drépanocytaires majeurs. L'enfant hérite d'un gène HbS d'un parent et d'un gène HbC de l'autre (hémoglobine C, mutation sur le même codon 6 de la chaîne bêta).
Particularités cliniques
Longtemps considérée comme "plus douce" que HbSS, la forme HbSC est aujourd'hui reconnue comme sévère à ne pas sous-estimer :
- Anémie moins profonde (taux d'hémoglobine souvent entre 10–12 g/dL vs 7–9 g/dL en HbSS)
- Mais viscosité sanguine plus élevée → risque accru de :
- Nécrose avasculaire de la tête fémorale
- Rétinopathie proliférative (risque plus élevé qu'en HbSS)
- Embolie pulmonaire
L'espérance de vie médiane est d'environ 54–57 ans, avec une survie à 18 ans de 98,4 % (vs 93,9 % en HbSS).
La rétinopathie et la nécrose de hanche doivent être surveillées de façon spécifique dans la forme HbSC, même chez des patients qui "se sentent bien".
HbS/β-thalassémie — Une sévérité variable
Cette forme résulte de l'association d'un gène HbS et d'une mutation de β-thalassémie. Deux sous-types existent selon le niveau de production résiduelle d'hémoglobine A normale :
HbS/β⁰-thalassémie
- Aucune production d'HbA normale
- Tableau clinique comparable à HbSS : sévère, avec crises vaso-occlusives fréquentes et hémoglobine basse
HbS/β⁺-thalassémie
- Production partielle d'HbA (15–30 % en général)
- Forme plus modérée : anémie moins marquée, complications souvent plus tardives
- L'électrophorèse de l'hémoglobine montre HbS + HbA + HbA2 élevée + HbF
Formes rares : HbSD, HbSO-Arab, HbSE
Ces formes composite sont rares (< 1 % chacune) mais cliniquement significatives :
- HbSD-Punjab : hétérozygote composite HbS/HbD. Tableau clinique sévère, comparable à HbSS dans certains cas. Prévalent en Asie du Sud (Inde, Pakistan).
- HbSO-Arab : forme sévère avec complications vaso-occlusives fréquentes. Décrite initialement dans les populations arabo-africaines.
- HbSE : généralement peu symptomatique, anémie discrète. HbE est fréquente en Asie du Sud-Est.
Récapitulatif comparatif
| Forme | Fréquence | Sévérité | Anémie | Espérance de vie |
|---|---|---|---|---|
| HbSS | ~70 % | Sévère | Profonde (7–9 g/dL) | ~48 ans |
| HbSC | ~25 % | Modérée à sévère | Légère (10–12 g/dL) | ~54 ans |
| HbS/β⁰ | ~4 % | Comparable à HbSS | Profonde | ~48 ans |
| HbS/β⁺ | ~1 % | Modérée | Légère à modérée | Meilleure |
| HbSD, HbSO | < 1 % | Variable | Variable | Variable |
Le dépistage néonatal, clé de la prise en charge précoce
En France, le dépistage néonatal de la drépanocytose est systématique pour les nouveau-nés à risque depuis 1985 (étendu à l'ensemble du territoire en 2000 pour les populations à risque). Il repose sur l'électrophorèse de l'hémoglobine réalisée à J3, qui permet d'identifier la forme précise et d'initier rapidement :
- La prophylaxie antibiotique (pénicilline V dès 2 mois)
- La supplémentation en acide folique
- La surveillance spécialisée et l'éducation des parents
La connaissance de la forme génétique exacte est fondamentale car elle oriente la surveillance, le traitement de fond (hydroxyurée, échanges transfusionnels) et les indications de greffe de cellules souches hématopoïétiques.
Commentaires (3)
J'ai la forme HbSC et pendant longtemps on m'a dit que c'était "beaucoup moins grave". Cet article montre bien que ce n'est pas si simple et que le suivi reste nécessaire.
Enfin une explication claire sur la β-thalassémie associée à la drépanocytose. Mon médecin ne prenait pas le temps d'expliquer les différences entre les génotypes.
Ma fille a été diagnostiquée HbSS à la naissance grâce au dépistage néonatal. J'aurais aimé avoir cet article pour comprendre dès le début ce que ça signifiait vraiment.